





Abstract
In peripheral neurons, intracellular hyperglycemia associated with diabetes mellitus causes a redirection of carbohydrate flux from glycolysis to pathways that contribute to diabetic complications. For example, these pathways are responsible for pathological changes in peripheral neurons resulting in diabetic polyneuropathy. Shunting carbohydrate flux away from these complication-causing pathways reduces their pathological impact. The actions of transketolase (TK), a thiamine (vitamin B1)-dependent enzyme, shunts carbohydrate flux into the pentose phosphate pathway (PPP) away from these complication-causing pathways. Diabetics have been shown to be endemically thiamine-deficient. High-dose thiamine therapy reduces flux through these complication-causing pathways. Benfotiamine, a highly bioactive thiamine derivative, blocks the pathological impact of these complication-causing pathways and activates TK by tenfold compared to thiamine. Benfotiamine has also been shown to diminish the intensity of the symptoms of diabetic polyneuropathy. Thiamine deficiency is a well-established cause of polyneuropathy. These facts suggest a strong interrelationship between thiamine deficiency and the polyneuropathy of diabetes.
The Case for Thiamine Deficiency in Diabetics
Saito et al showed that blood thiamine levels were below normal in 76.1% of elderly diabetic outpatients who had not received previous thiamine supplementation.1 Valerio et al found the plasma content of thiamine in 10 type I diabetic children was 35 nmol/L vs 53 nmol/L in 6 age-matched nondiabetic control subjects.2 This agrees with the work of Haugen who found lower thiamine levels in children with diabetes.3
Jermendy looked at the erythrocyte transketolase activity (ETA) in 75 patients with diabetes and 60 healthy subjects without diabetes.4 Erythrocyte transketolase activity is a measure of transketolase (TK) activity in red blood cells before and after saturation with exogenous thiamine; the higher the ETA, the higher the risk for thiamine deficiency. The ETA of patients with diabetes was significantly higher than that of healthy, nondiabetic subjects (1.14±0.01 vs 1.08±0.02; P<0.01), indicating the diabetics tested were at greater risk for thiamine deficiency compared to the nondiabetic controls. Havivi et al found the percentage of diabetic patients with thiamine deficiency was significantly higher than the healthy population.5 Thornalley et al measured plasma thiamine status, including free thiamine, thiamine monophosphate, and thiamine diphosphate levels in type I and type II diabetics vs nondiabetic healthy volunteers.6 Plasma thiamine concentration was decreased 76% and 75% in type 1 and type 2 diabetic patients, respectively. The plasma thiamine status was 64.1±12.0 nmol/L in normal volunteers, 15.3±9.6 nmol/L in patients with type 1 diabetes, and 16.3±11.5 nmol/L in patients with type 2 diabetes (mean±SD; P<0.001). Renal clearance of thiamine was increased 24-fold in patients with type 1 diabetes and 16-fold in patients with type 2. These results strongly suggest that kidney dysfunction is a major contributing factor in the endemic thiamine-deficient state associated with diabetics.
Similarities in the Clinical Characteristics of Diabetic Polyneuropathy and Thiamine Deficiency–Induced Polyneuropathy
Peripheral neuropathy afflicts 60% to 70% of diabetic patients.7 The most common type of neuropathy experienced by diabetics is a distal sensorimotor polyneuropathy (DSP). The Rochester Diabetes Project found that 72% of all neuropathies experienced by patients with type 2 diabetes mellitus were of this type.7 It is a distal symmetrical neuropathy of axonal degeneration characterized by pain, paresthesia, hyperesthesia, dysesthesia, proprioceptive defect, decreased sensation, muscle weakness, and atrophy. It is known to progress in a stocking-glove distribution, although its presentation can be highly variable.8-12
Thiamine deficiency also causes a DSP characterized by axonal degeneration.13 Its clinical presentation is often similar to diabetic DSP.14 Clinicians have long noted the similarities in the characteristics of diabetic DSP and thiamine deficiency–induced DSP.5,15 The parallels in the clinical presentation of both neuropathies were thought to be so remarkably similar that in 1961 attempts were made, albeit unsuccessfully, to demonstrate a correlation between excessive thiamine urinary excretion in diabetics and the classical symptom of polyuria commonly seen in diabetics.5
Thiamine and Benfotiamine
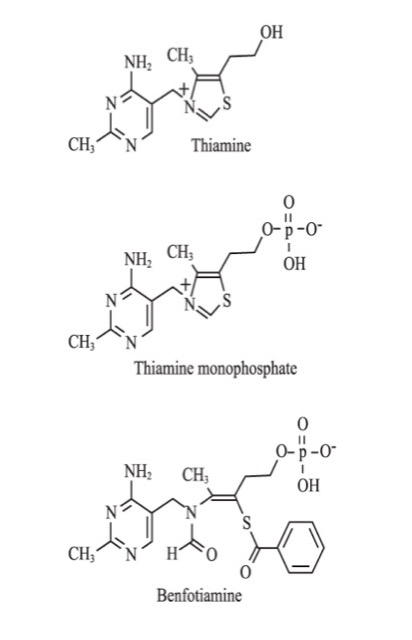
Thiamine, also known as vitamin B1, exists in the human body as free thiamine and its phosphate esters: thiamine monophosphate, thiamine pyrophosphate (TPP), and thiamine triphosphate (Figure 1).16 Thiamine pyrophosphate acts as an essential coenzyme for pyruvate dehydrogenase (PDH), α-ketoglutarate dehydrogenase (αKGDH), branched chain keto-acid dehydrogenase, and TK. These enzymes are essential in the metabolism of carbohydrates.17,18 Thiamine also plays a role in the biosynthesis of nucleic acids and neurotransmitters.17-19 It has a role as a modulator of neuronal and neuromuscular transmission, probably through ionic channel regulation.20,21 Thiamine has been shown to be an antioxidant.22-25
In addition to causing a DSP, gross thiamine deficiency is a well-known cause of heart failure (wet beriberi), encephalopathy, and psychosis (Wernicke-Korsakoff syndrome).14,26 Thiamine deficiency has also been observed to cause DNA fragmentation in cultured neurons and impaired neuronal energy metabolism.17,27 Thiamine deficiency has been shown to kill neurons but not nonneuronal cells in culture.27
Thiamine is water-soluble.26 Absorption from the gut takes place mainly from the proximal small intestine primarily via an energy-dependent active transport system.28 Only about 5% of ingested thiamine is absorbed.29
Benfotiamine is the most well-studied and most commonly used lipid-soluble thiamine derivative (Figure 1). Wada et al at the Takamine Laboratory of the Sankyo Company of Japan developed benfotiamine in the early 1960s as a treatment for symptomatic thiamine deficiency.30 It has been used in Germany since 1985 and subsequently in many other countries for the treatment of diabetic polyneuropathy.31
Benfotiamine is absorbed from the gut via passive diffusion and converted into thiamine monophosphate after absorption. In 1996 Schreeb et al measured plasma thiamine levels after subjects ingested equimolar quantities of either thiamine mononitrate, a commonly used salt of thiamine, or benfotiamine. The group ingesting benfotiamine had maximum plasma thiamine levels that were 6.7 times higher than the group ingesting thiamine mononitrate.32
Both thiamine and benfotiamine are known activators of TK. Benfotiamine has been shown to activate TK in type I diabetics by approximately 250%, while thiamine has been shown to activate TK by approximately 25% in thiamine-deficient subjects.33-35
Figure 1. The Structure of Thiamine, Thiamine Monophosphate, and Benfotiamine
Thiamine Deficiency and Insulin
Insulin and thiamine have been shown in animal models to have a regulatory effect on each other.36 Rathanaswami et al showed that thiamine deficiency decreases insulin release in beta islet cells and impairs the biosynthesis of insulin.37,38 Patrini et al showed that low levels of insulin reduce thiamine absorption from the gut.39
Cellular Respiration and the Pentose Phosphate Pathway
In human cells, energy is generated through cellular respiration, a metabolic process involving glycolysis, the citric acid cycle, and the mitochondrial electron transport chain. Cellular respiration is an aerobic process in which energy is extracted from glucose and converted into adenosine triphosphate (ATP).40
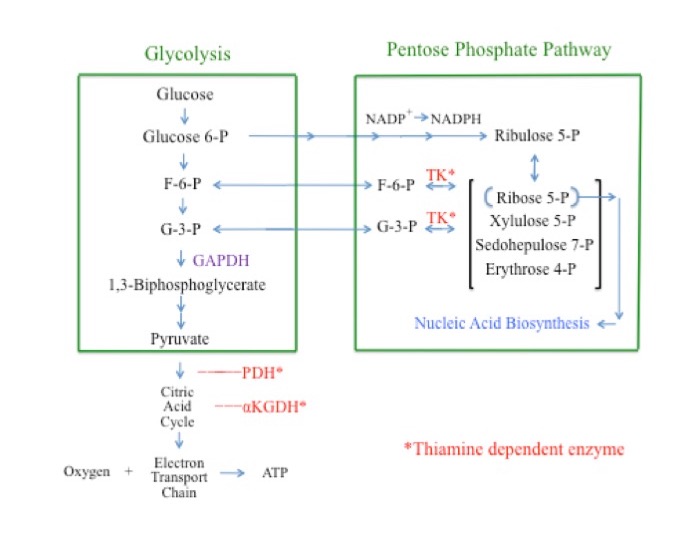
Glycolysis is a cytosolic pathway that extracts energy from glucose via its conversion to pyruvate.41 In the first steps of glycolysis, glucose is converted to glucose 6-phosphate (G6P), then on to fructose 6-phosphate (F6P), fructose 1,6-biphosphate, and glyceraldehyde 3-phosphate (G3P). Glyceraldehyde 3-phosphate is then converted to 1,3-biphosphoglycerate through the actions of the enzyme glyceraldehyde phosphate dehydrogenase (GAPDH). Inhibition of GAPDH consequently results in an increased concentration of “upstream” glycolytic intermediaries, including glucose, G6P, F6P, and G3P. Pyruvate is the end product of glycolysis. Pyruvate and its subsequent metabolites are further metabolized in the mitochondria through the citric acid cycle and the electron transport chain for additional energy production (Figure 2).42-44
Thus, there appears to be a unifying mechanism in the pathobiology of diabetic complications: superoxides formed in cells with increased intracellular glucose levels cause the activation of multiple destructive signaling pathways.
The pentose phosphate pathway (PPP) is a cytosolic pathway. It produces, among other carbohydrates, ribose 5-phosphate, an essential sugar for the manufacture of nucleic acids. The PPP is the cell’s primary source for the production of the important reducing agent NADPH.45-48 The PPP also produces G3P and F6P. As such, G3P and F6P are metabolites of both glycolysis and the PPP. The thiamine-dependent enzyme TK is a rate-limiting enzyme of the PPP. The direction of the TK reaction is driven by substrate concentrations. Under normal physiologic conditions the glycolytic intermediaries F6P and G3P are end products of the TK reaction and flow from the PPP into glycolysis. Under conditions in which the concentrations of G3P and F6P are greatly elevated, as is the case when GAPDH is inhibited, the direction of the TK reaction reverses, shunting G3P and F6P away from glycolysis and into the PPP.49
Figure 2. Simplified Overview of Aerobic Cellular Respiration and the Pentose Phosphate Pathway
Oxidative Stress
An imbalance between oxidants and antioxidants in favor of the oxidants, potentially leading to damage, is termed oxidative stress. Oxidants are formed as a normal product of aerobic metabolism but can be produced at elevated rates under pathophysiological conditions.50 Reactive oxygen species (ROS) include oxygen ions, superoxides, free radicals, and peroxides. They are generally very small molecules that are highly reactive due to the presence of unpaired valence shell electrons. High levels of ROS cause significant damage to DNA, enzymes, lipids, proteins, and other macromolecules with subsequent damage to cellular processes.51 Oxidative stress contributes to a number of human disorders such as diabetes, cancer, cardiovascular diseases, stroke, and late-onset neurodegenerative disorders. It also contributes to the aging process.45,52 Oxidative stress is responsible for abnormalities in thiamine-dependent processes and has been shown to convert thiamine to biologically inactive forms.17,23,24
Oxidative Stress, Thiamine Deficiency, and Diabetes
Thiamine deficiency induces elevated levels of oxidative stress in humans with an accompanying pathological impact on cellular functions.53-55 The thiamine-dependent enzymes PDH and α-ketoglutarate dehydrogenase complex (αKGDH ) are sensitive to and partially inactivated by oxidative stress, further magnifying the negative impact of thiamine deficiency on thiamine-dependent cellular processes.17
Oxidative stress induced by elevated intracellular glucose levels is a well-established etiologic factor in the pathogenesis of diabetic complications.34,54,56-60 Under normoglycemic conditions, energy is extracted from glucose in a precisely regulated manner via glycolysis, the citric acid cycle, and the mitochondrial electron transport chain.34,54 During episodes of hyperglycemia, most cell types are able to maintain their intracellular glucose levels within normal physiologic limits by limiting further influx of glucose. In contrast, hyperglycemia causes increased intracellular glucose levels within capillary endothelial cells of the retina, mesangial cells in the renal glomerulus, and neurons and Schwann cells in peripheral nerves. When intracellular glucose levels are pathologically high, there is increased flux through glycolysis, the citric acid cycle, and the electron transport chain. This increased flux increases intracellular oxidative stress via the over-production of superoxides by the electron transport chain. Under these circumstances, capillary endothelial cells of the retina, mesangial cells in the renal glomerulus, and neurons and Schwann cells in peripheral nerves are selectively damaged.54,61,62
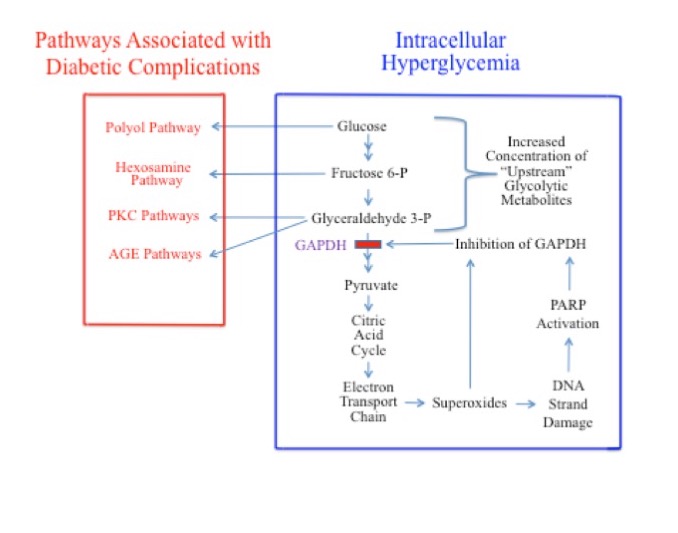
It is no coincidence that the tissues associated with these cell types are those that are most prone to diabetic complications. Brownlee et al has shown that hyperglycemia-induced superoxides are the cause of many complications associated with diabetes.54 Superoxides directly inhibit the activity of the glycolytic enzyme GAPDH and cause DNA strand damage. DNA strand damage induces the production of poly(ADP-ribose)polymerase (PARP), which, in addition to being involved in DNA repair, further inhibits the activity of GAPDH (Figure 3). These processes may inhibit GAPDH by as much as 66%. Inhibition of GADPH causes an accumulation of the “upstream” glycolytic metabolites, glucose, G6P, F6P, and G3P.34,54,61,62 This vicious cycle then goes on to wreak further damage.
The increased concentration of glucose, F6P, and G3P in the cytosol, secondary to GAPDH inhibition, activates 4 signaling pathways known to be responsible for diabetic pathobiology on the metabolic level: 1) the polyol pathway (activated by increased glucose);63,64 2) the hexosamine pathway (activated by increased F6P);65,66 3) pathways responsible for increased formation of advanced glycation end products (AGEs; activated by increased G3P); and 4) pathways responsible for increased formation of protein kinase C (PKC) isoforms (also activated by G3P).54
Figure 3. GAPDH Inhibition During Elevated Intracellular Glucose Levels and Resultant Activation of Signaling Pathways Responsible for Complications Associated with Diabetes
Thus, there appears to be a unifying mechanism in the pathobiology of diabetic complications: superoxides formed in cells with increased intracellular glucose levels cause the activation of multiple destructive signaling pathways. The discovery of this unified mechanism represents a significant advance in the understanding of the etiology of diabetic complications.34,54,62,66 Additionally, Brownlee et al have shown that hyperglycemia-induced ROS increase the expression of AGE receptors and their ligands thus further contributing to this pathobiology.67
Benfotiamine and High-Dose Thiamine Counteract Oxidative Stress
Increased flux through the PPP counteracts oxidative stress by increasing the production of NADPH (nicotinamide adenine dinucleotide phosphate hydrogen).52 Benfotiamine has been shown to raise flux through the PPP.68 Schmid et al and Katare et al have demonstrated that benfotiamine exhibits direct antioxidative capacity.70,69 Thiamine has been shown to be an antioxidant.22-24,70
Thiamine and Benfotiamine Reduce the Impact of Pathways Responsible for Diabetic Pathobiology
Benfotiamine has been shown to block 4 signaling pathways associated with diabetic complications. In 2003 Brownlee et al showed that benfotiamine blocks the hexosamine, AGE, and PKC pathways in experimental diabetic retinopathy by activating the PPP enzyme TK.71 In 2006, Porta et al showed that by the same mechanism benfotiamine and thiamine correct polyol pathway activation induced by high glucose in vascular cells.66
Thiamine therapy has been shown to help counter diabetic complications and blocking pathways associated with these complications. Thiamine therapy has been shown to decrease hexosamine pathway activity, polyol pathway activity, PKC activation, AGE formation, and oxidative stress.66,72-76 Thiamine has also been shown to reverse hyperglycemia-induced dysfunction in cultured endothelial cells and improve glycemic control.77,78
Benfotiamine has been shown in multiple studies to reduce the symptoms of diabetic polyneuropathy. In 2008 Stracke et al demonstrated the safety and efficacy of benfotiamine in a 6-week double-blind, placebo-controlled, phase III trial of 124 patients with symmetrical, distal, diabetic polyneuropathy (P=0.033).79 In 2003 Haupt et al published the results of a 3-week randomized controlled pilot study of benfotiamine in 40 patients with diabetic polyneuropathy. The study showed a statistically significant improvement in neuropathy score in diabetics with polyneuropathy treated with benfotiamine vs controls (P=0.0287).80 In 1999 Winkler et al demonstrated the effectiveness of benfotiamine in the treatment of painful diabetic neuropathy; improvement was greatest in the group given the highest dose.81 Brownlee et al showed that benfotiamine in combination with the antioxidant alpha-lipoic acid normalized markers of ROS-induced complication-causing pathways in type I diabetes.35
Discussion
Oxidative stress is produced by both thiamine deficiency and intracellular hyperglycemia. During episodes of oxidative stress the glycolytic enzyme GAPDH is inhibited. Inhibition of GAPDH increases concentrations of the “upstream” glycolytic metabolites: glucose; G6P; F6P; and G3P. This results in the shunting of glucose, F6P, and G3P from glycolysis into signaling pathways associated with diabetic complications and the shunting of G6P into the PPP, generating increased production of the reducing agent NADPH.
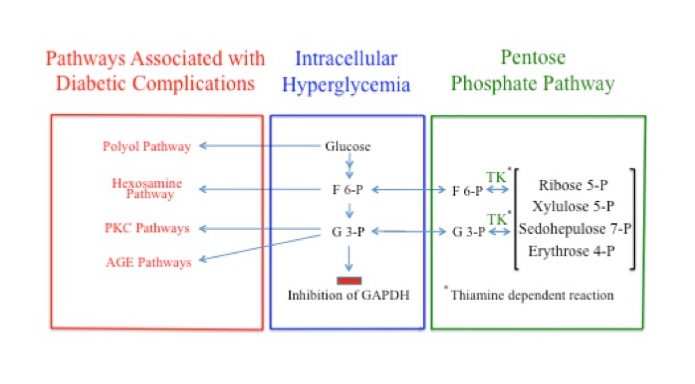
Glyceraldehyde 3-phosphate and F6P are produced by both the PPP and glycolysis. Under normal physiological concentrations, G3P and F6P are products of the enzymatic actions of TK and flow from the PPP into glycolysis. But when the concentrations of G3P and F6P are sufficiently elevated, as when GAPDH is inhibited, the direction of the TK reaction reverses, redirecting G3P and F6P from glycolysis into the PPP where they are converted into other sugars. The flow of G3P and F6P into the PPP under these conditions is dependent on adequate levels of thiamine, via its action as a coenzyme for TK. Consequently, thiamine deficiency, by reducing flux through TK, likely increases the pathological impact of the signaling pathways responsible for complications associated with diabetes. In the case of peripheral neurons and Schwann cells, this would impair peripheral nervous system function and intensify the symptoms of diabetic polyneuropathy (Figure 4).
Figure 4. Multiple Possible Pathways of Fructose 6-Phosphate and Glyceraldehyde 3-Phosphate During GAPDH Inhibition
This interrelationship appears to be validated by evidence that benfotiamine, a strong activator of the thiamine-dependent enzyme TK, has been shown to reduce the symptoms of diabetic polyneuropathy and that both benfotiamine and thiamine have been shown to reduce the pathological impact of these complication-causing pathways associated with diabetic pathobiology.
Other complications of diabetes attributed to increased flux through these 4 signaling pathways, such as retinopathy, microvascular disease, and nephropathy, have also been shown to improve when treated with either benfotiamine or thiamine.73,77,82-89 Additionally, diabetes mellitus is a clinical component of thiamine-responsive megaloblastic anemia syndrome, a disease characterized by cellular thiamine processing defects.90 Peripheral nerve dysfunction in the presence of thiamine deficiency is consistent with evidence that abnormal thiamine-dependent processing is related to the central neuronal dysfunction observed in Alzheimer’s disease.91,92
The preponderance of evidence would indicate that diabetics have an increased need for thiamine while at the same time being endemically deficient in it. Perhaps Porta et al stated it best:
Diabetes might be considered a thiamine-deficient state, if not in absolute terms at least relative to the increased requirements deriving from accelerated and amplified glucose metabolism in non-insulin dependent tissues that, like the vessel wall, are prone to complications.21
When taken as a whole, the evidence suggests a significant interrelationship between thiamine deficiency and diabetic polyneuropathy.
Conflict of Interest Disclosure
The author is the founder and CEO of Realm Labs, LLC, a distributor of vitamin supplements, including supplements containing benfotiamine.
