





Abstract
EPs 7630 is a proprietary extract from the roots of the Pelargonium sidoides plant. The herb has long been used to treat cough, sore throat, congestion, and other respiratory ailments. The EPs 7630 extract has been the subject of 20 clinical studies involving more than 9,000 patients, including 3,900 children. It has been shown to safely and effectively treat acute upper respiratory tract infections such as bronchitis, tonsillopharyngitis, sinusitis, and the common cold. The extract is also supported by safety data from postmarketing surveillance studies and pharmacovigilance based on use in Germany and elsewhere.
Introduction
A member of the Geraniaceae family, P sidoides DC is native to the coastal regions of South Africa.1 The plant is notable for its narrow, deep-red flowers and its large, heart-shaped leaves. Along with the closely related P reniforme Curt, the root has been used for centuries as a traditional herbal remedy in South Africa.2 In the late 19th century, a product made from the root, “Stevens’ Consumption Cure,” gained some popularity in England as a cure for tuberculosis. In the 1920s, Dr A Sechehaye reportedly treated approximately 800 tuberculosis patients with a preparation of the root and published case studies.2



Pelargonium sidoides
In 1972, German researchers discovered the chemical profile identity of the P sidoides root.3 As research progressed, a proprietary extraction technique was developed and perfected to yield EPs 7630. It was determined that 3-year-old rhizomes contain an optimal amount of active constituents. Currently, P sidoides is grown on specialized farms in South Africa using ecological cultivation methods. An ethanolic root extract (1:9-11), EPs 7630 is manufactured by Dr Willmar Schwabe Pharmaceuticals, Karlsruhe, Germany, and registered by ISO Pharmaceuticals, Ettlingen, Germany.
Phytochemistry and Pharmacology
The EPs 7630 extract contains primarily polyphenols (mainly catechin and gallocatechin), proteins, minerals, and, in lower concentrations, 7-hydroxycoumarin derivatives (Figure 1).4 These 7-hydroxycoumarin (including umckalin) derivatives differ in chemical structure from the known anticoagulant coumarins and are not associated with anticoagulant activity or interaction with warfarin and its pharmacokinetics.5
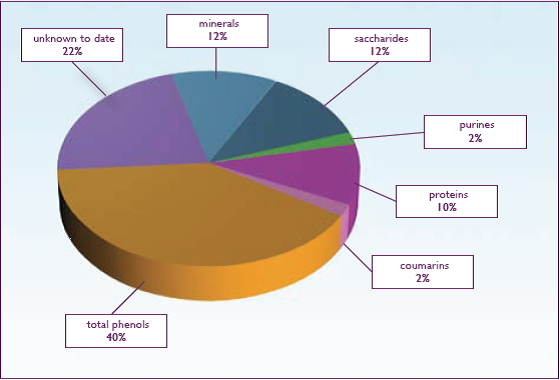
Figure 1.Chemical Composition of EPs 7630
Pharmacological studies have suggested that the mechanism of action of EPs 7630 is multifactorial.6 In vitro studies have found that it exerts a cytoprotective effect against virus-induced cell destruction and also increases release of antimicrobial peptides (also know as defensins) from neutrophilic granulocytes.7,8 Studies have also found that EPs 7630 acts as an immune stimulant. These actions are mediated mainly by the release of tumornecrosis factor (TNF-a) and nitric oxide, stimulation of interferon-b synthesis, and increase of natural killer cell activity.9 Studies have also found that EPs 7630 increases phagocytosis.10
While EPs 7630 is not considered antibacterial, in vitro studies have suggested that it inhibits bacterial adhesion to epithelial cells.11 Finally, EPs 7630 has also been found to stimulate ciliary beat frequency in vitro.12 This action may support a mucolytic (secromotoric) effect during acute upper respiratory tract infections.
Clinical Research
EPs 7630 has been shown to reduce the duration and severity of acute upper respiratory tract infections, including bronchitis, tonsillopharyngitis, sinusitis, and the common cold. A 2008 meta-analysis was published by the Cochrane Review and includes some of the studies reviewed in the following sections.13 The efficacy and safety of EPs 7630 in the treatment of acute upper respiratory tract infections has been studied in over 3,800 patients in controlled double-blind studies and over 5,400 patients in open-label and noninterventional (postmarketing surveillance) studies. The safety and efficacy data includes children as young as 1 year.
Acute Bronchitis
A meta-analysis published in 2008 reviewed 6 randomized, controlled clinical studies demonstrating the safety and efficacy of EPs 7630 for the treatment of acute bronchitis in both adults and children.14 While one study compared EPs 7630 against N-acetylcysteine, the other 5 were placebo-controlled trials. All studies treated patients for 7 days. The primary outcome measure was the Bronchitis Severity Score (BSS). The BSS measures the following features of acute bronchitis: cough, sputum, rales/rhonchi, chest pain during coughing, and dyspnea. Each symptom was scored by an investigator using a 5-point rating scale ranging from 0 to 4 (0=absent; 1=mild; 2=moderate; 3=severe; 4=very severe). All 6 studies demonstrated a significant decrease in the BSS after 7 days of treatment with EPs 7630. The review’s authors concluded, “Currently available data from 6 high quality randomized clinical trials suggests there is encouraging evidence that P sidoides is effective compared to placebo for patients with acute bronchitis.” Three of these clinical trials are detailed below.
In one study, 468 adult male and female subjects (age≥18 years) diagnosed with acute bronchitis ≤48 hours and having a BSS≥ 5 points were recruited for a randomized, double-blind, placebo-controlled trial.15 Patients received either EPs 7630 or a placebo at a dose of 1.5 mL 3 times per day for 7 days. In the case of fever (≥39° C), acetaminophen tablets (500 mg) were allowed.
The primary outcome measure was the change in the BSS on day 7 in relation to the baseline score. Secondary outcome measures included: (1) prospective defined response criteria based on the BSS (A: BSS<3 points; B: decrease of BSS≥7 points; C: A+B); (2) treatment outcome according to the Integrative Medicine Outcome Scale (IMOS); (3) onset of treatment effect; (4) consumption of paracetamol (acetaminophen); (5) change of individual symptoms of BSS and further symptoms; (6) patients’ health status using health-related quality of life questionnaires (SF-12 Health Survey, EQ-5D); and (7) questions about the complaints and satisfaction with treatment using the Integrative Medicine Patient Satisfaction Scale (IMPSS). Safety was assessed with respect to frequency, nature, and severity of adverse events. Tolerability of treatment was also assessed by investigators and patients, as well as by laboratory tests. After enrollment (day 0), examinations occurred on day 3, 4, or 5, and on day 7. At each visit the investigator recorded clinical status, reviewed the patient’s diary, documented the consumption of acetaminophen and recorded the number of adverse events. On day 7, there was a final assessment, which included laboratory tests and sputum analysis.
On day 7, BSS had decreased by 5.9±2.9 in the EPs 7630 group and by 3.2±4.1 in the placebo group compared to baseline (Figure 2). The 95% confidence interval (CI) for the difference of effects between the two treatment groups (EPs 7630 minus placebo) was calculated as [–3.359; –2.060], showing a significant superiority of EPs 7630 over the placebo by day 7 (P<.0001). This statistically significant difference was observed as early as the first follow-up visit (day 3, 4, and 5) with a BSS score of 4.8±2.3 points in the EPs 7630 group compared with 6.2±3.0 in the placebo group (P<.0001). In patients with the highest BSS at baseline (defined as a BSS≥8 points), there was a statistically significant decrease in the BSS in the EPs 7630 group (6.8±2.7) compared with the placebo group (4.5±4.2) at day 7 (P<.0001). A BSS of <3 points (response criteria A) was observed in 64% of patients in the EPs 7630 group compared with 37.9% in the placebo group (P<.0001). A decrease in BSS of at least 7 points (response criteria B) was observed in 43.3% of patients in the EPs 7630 group compared with 23.0% of patients treated with the placebo at day 7 (P<.0001). Rapid recovery (defined as response criteria C) was observed in 34.3% of EPs 7630 patients compared with 20.4% receiving the placebo (P<.0001).
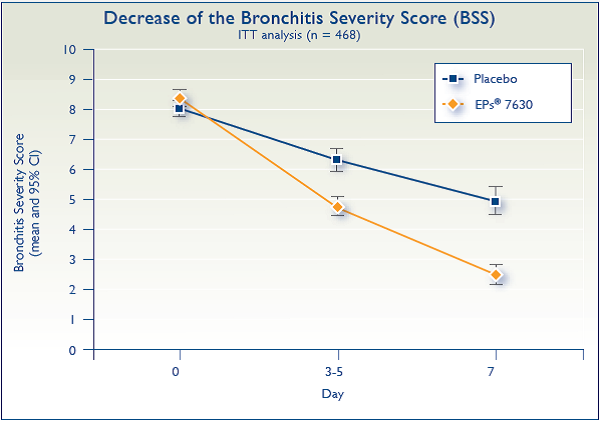
Figure 2.
A statistically significant improvement occurred in the individual symptoms of rales/rhonchi and chest pain during coughing in the EPs 7630 group compared with the placebo group (P<.0001). In the EPs 7630 group, cough disappeared or improved in 89.2% of patients compared with 56.6% of patients in the placebo group (P<.0001), and sputum disappeared or improved in 66% of patients in the EPs 7630 group compared with 47.7% of those in the placebo group (P<.0002). On day 7, fever had disappeared in 96.9% of patients in the EPs 7630 group compared with 58.4% of those in the placebo group (P<.0001). Patients in the EPs 7630 group were able to return to work nearly two days earlier than the placebo-treated group (P<.0001). Adverse events were mild and similar in both groups: 8.6% in the EPs 7630 group and 6.8% in the placebo group. These events included ear-nose-throat (ENT) and respiratory complaints (likely due to the existing condition), as well as mild gastrointestinal upset.
A second randomized, double-blind, placebo-controlled trial studied the effect of EPs 7630 in 124 adults (≥18 years old) with acute bronchitis (duration of complaints ≤48 hours).16 Patients were required to have a BSS of ≥5 points. Patients received either EPs 7630 or a placebo at a dose of 1.5 mL three times per day for 7 days. Patients were instructed to take test medication 30 minutes before or after meals. In the case of fever (≥39° C), acetaminophen tablets (500 mg) were allowed.
The primary outcome was the change in the BSS at day 7. Secondary outcome measures were as follows: BSS<5 points at the end of the study; decrease of BSS of ≥5 points between baseline and the end of the study; onset of treatment effect; consumption of acetaminophen; change of individual symptoms of BSS; patients’ health status using the health-related quality of life questionnaires (SF-12 Health Survey, EQ-5D). Treatment outcome was also measured by both the patient and investigator using the Integrative Medicine Outcome Scale (IMOS), which consists of a 5-point rating scale (complete recovery; major improvement; slight to moderate improvement; no change; deterioration). Satisfaction with treatment was measured using the Integrative Medicine Patient Satisfaction Scale (IMPSS), another 5-point scale ranging from “very satisfied” to “very dissatisfied.”
Treatment outcome was also measured by both the patient and investigator using the Integrative Medicine Outcome Scale (IMOS), which consists of a 5-point rating scale.
Although all 124 patients were included in the intention-to-treat analysis, 3 patients in the EPs 7630 group and 4 patients in the placebo group had dropped out by day 7. On day 7, BSS decreased by 7.2±3.1 points in the EPs 7630 group compared with 4.9±2.7 points in the placebo group (P<.0001) (Figure 3). The greater treatment effect was noted at the first followup contact day after baseline (either day 3, 4, or 5) with a BSS of 4.4±2.2 points in the EPs 7630 group and 6.2±2.5 points in the placebo group (P<.0001). A BSS of less than 5 points was observed in 61 of 64 patients (95.3% in the EPs 7630 group compared with 35 of 60 patients (58.3%) in the placebo group (P<.0001). A decrease of at least 5 points compared with baseline was seen in 58 of 64 patients (90.6%) in the EPs 7630 group and 31 of 60 patients (51.7%) in the placebo group (P<.0001). Rapid recovery, defined as fulfillment of both of the above outcomes, was observed in 58 of 64 patients in the EPs 7630 group compared with 25 of 60 patients in the placebo group (P<.0001). On day 7, rate of complete recovery from individual symptoms on the BSS was considerably higher in the EPs 7630 group. On day 7, rales/rhonchi had disappeared in 55 of 60 patients (91.7%) in the EPs 7630 group compared with 29 of 59 patients (49.2%) in the placebo group (P<.0001) and chest pain during coughing had disap- peared in 55 of 58 patients (94.8%) in the EPs 7630 group and 29 of 52 (55.8%) in the placebo group (P<.0001). Among the 5 symptoms, cough had the slowest recovery in both groups but was significantly greater in the EPs 7630 group—31.3 % in the EPs group and 5.0% in the placebo group (P<.0001).
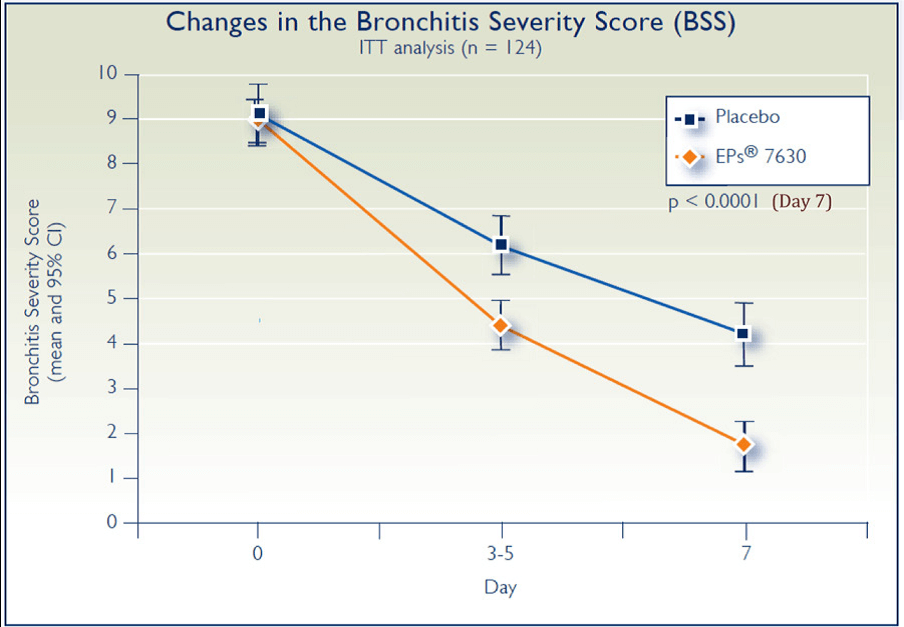
Figure 3.
On day 7, the IMOS results showed that 54 of 64 patients (84.4%) in the treatment group were assessed as having major improvement or complete recovery compared with 18 of 60 patients (30.0%) in the placebo group. Similar differences were also noted for the EQ-5D health-related quality of life scale as well as IPSS satisfaction with treatment scale. Tolerability of treatment was very good or good in both groups and there were a total of 15 patients in the EPs 7630 group and 10 in the placebo group who reported adverse events during the trial. All adverse events were assessed as being not serious.
Finally, in a third randomized, double-blind, placebo-controlled study, 217 adults (18 to 66 years old) with acute bronchitis (duration of complaints ≤48 hours) were randomized to receive either 1.5 mL of EPs 7630 or placebo three times per day for 7 days.17 Patients were required to have a BSS of ≥5 points. The primary outcome was the change in the BSS at day 7. Secondary outcome measures included changes in general symptoms such as hoarseness, headache, pain in the limbs, fatigue, and fever. Patients were asked to rank their satisfaction with treatment using the IMPSS. Investigators assessed efficacy using the IMOS. All outcome measures were completed at baseline, the first follow-up visit (from day 3 to 5), and at the end of treatment (day 7).
At day 0, the BSS was 8.9±1.6 points for the treatment group and 8.4±1.8 points for the placebo group. At the first followup visit, the BSS had decreased to 4.2±2.0 points in the treatment group compared with 5.9±2.5 points in the placebo group. By day 7, the BSS was 1.3±2.1 points in the treatment group compared with 3.1±3.0 points in the placebo group. Between baseline and day 7, the BSS decrease in the EPs 7630 group was 7.6±2.2 points and 5.3±3.2 points in the placebo group (P<.0001) (Figure 4). On the IMOS ratings, 45.4% of patients taking EPs 7630 were assessed as having a complete recovery on day 7 compared with 6.4% in the placebo group. On the IMPSS rating, 89.8% of patients in the active treatment group reported complete recovery compared with 65.1% in the placebo group. In the active treatment group, 92.6% of patients reported complete remission or marked improvement in cough compared with 86.2% in the placebo group. Similar differences were noted for the other symptoms monitored. Adverse events were reported in 23 patients (21.3%) in the EPs 7630 group compared with 24 patients in the placebo group (22.0%). None was considered serious.
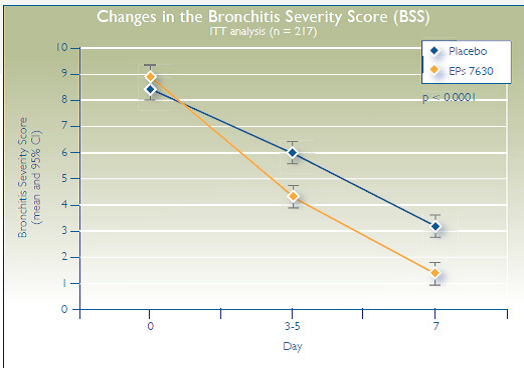
Figure 4.
Tonsillopharyngitis
In a randomized, double-blind, placebo-controlled trial, 143 children ages 6 to 10 years, with non–group A beta-hemolytic streptococcus (non-GABHS) tonsillopharyngitis received either EPs 7630 or a placebo at a dose of 1 mL three times per day for 6 days.18 Children enrolled in the study were diagnosed less than 48 hours before starting the trial with a negative rapid test for GABHS and a Tonsillopharyngitis Severity Score (TSS)≥8 points. In the case of fever (≥38.5° C), acetaminophen suppositories (500 mg) were allowed from day 0 to day 4. The main outcome measure was a change in TSS from baseline (day 0) to day 4. TSS measures two subjective features of acute tonsillopharyngitis: sore throat and functional impairment (difficulty swallowing). It also objectively measures symptoms of inflammation: pharyngeal erythema and fever. Each symptom was assessed by an investigator using a 4-point rating scale ranging from 0 to 3 (0=absent; 1=mild; 2=moderate; 3=severe). Secondary outcome criteria included: (1) response criteria based on the TSS; (2) change of individual symptoms and further complaints including headache; (3) treatment outcomes according to the IMOS (complete recovery, major improvement, slight to moderate improvement, no change, deterioration); and (4) patient activity level. After the enrollment day (day 0), controlled examinations occurred on days 2, 4, and 6. At each visit the investigator recorded clinical status, reviewed the patient’s diary, documented the consumption of acetaminophen, and recorded information about adverse events.
A statistically significant decrease occurred in the primary outcome criteria (change in TSS from day 0 to day 4) in the EPs 7360 group compared with the placebo group. The decrease of TSS from baseline (day 0) to day 4 was 7.0±2.4 points in the EPs 7630 group and 2.9±2.4 points in the placebo group (P<.0001). On day 2, TSS decreased from 10.3±1.2 to 6.8±2.2 in the EPs 7630 group compared with 9.7±1.4 to 8.2±2.8 in the placebo group (P<.0001), suggesting an early response in the EPS 7630 group.
A TSS of <5 points on day 4 was seen in 76.7% of patients in the EPs 7630 group compared with 34.3% of subjects in the placebo group (P<.0001) (Figure 5). A decrease of at least 5 points by day 4 was seen in 91.8% in the EPs 7630 group compared with 35.7% in the placebo group. Rapid recovery, defined as fulfillment of secondary response criteria 1 and 2, was observed in 75.3% of the EPs 7630 group and 32.9% of the placebo group (P<.0001). There was also an improvement seen in the activity level of subjects in the EPs 7630 group, but not in the placebo group. By day 6, the number of patients returning to school was 80.8% in the EPs 7630 group compared with 21.4% in the placebo group (P<.0001). Subjects in the EPs 7630 group consumed less acetaminophen than did subjects in the placebo group. No serious adverse events were reported.
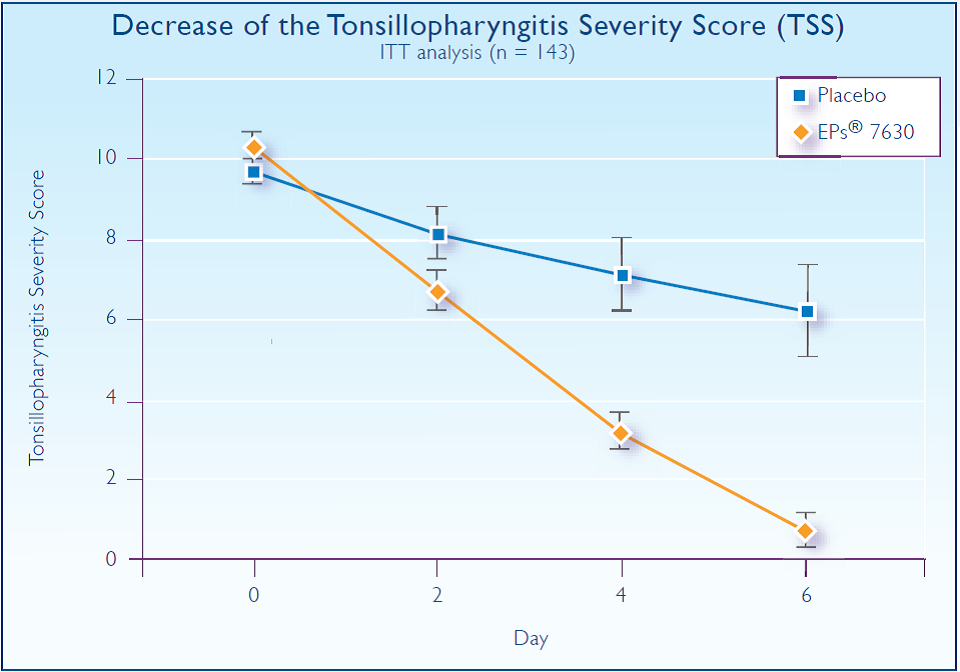
Figure 5.
Sinusitis
In a multicenter, prospective, randomized, double-blind, placebo- controlled study, 103 adult patients (18-60 y) with acute sinusitis were studied for 21 days.19 Patients recruited for the study had radiographically confirmed acute rhinosinusitis and a Sinusitis Severity Score (SSS) of ≥12 points. The SSS measures the following symptoms: 1) headache; 2) maxillary pain; 3) maxillary pain worse on bending forward, percussion, or pressure; 4) nasal obstruction; 5) purulent nasal secretion; and 6) purulent nasal discharge. The dose of EPs 7630 or placebo was 3 mL three times per day. The primary outcome measure was the change in SSS at day 7 of treatment compared to baseline. Secondary outcome criteria included response defined as a) an SSS<10 points on day 7, b) a reduction of at least 4 points on day 7, or c) both of the above. Other secondary outcomes included the occurrence of complete remission or substantial improvement of individual signs and symptoms as defined above; radiographic cure (“normal”) or substantial improvement at day 21; health-related quality of life as assessed on the 100 mm EQ-visual analogue scale (EQ-VAS) on day 7 (0=worst state of health, 100=best state of health); activity level; ability to work or engage in usual activities on day 7; and treatment outcome as assessed by the patient and investigator in the IMOS.
The decrease in SSS at day 7 was 5.5 points in the EPs 7630 group compared with 3.0 points in the placebo group (P<.0001) (Figure 6). This resulted in a between-groups difference of 3.0 points (95% CI; 2.0 to 3.9). By day 21, sinus x-rays were normal in more than 90% of the EPs 7630 group compared with 10% for the placebo group. This was most notable for the maxillary sinus with a nonsignificant trend for the frontal and ethmoid sinus. The mean improvement in the EQ-VAS was 13 mm higher for the EPs 7630 group versus placebo at day 7 (P<.0001). By day 7, 63% of the EPs 7630 group were back to work compared with 37% in the placebo group. Using the IMOS, the investigators assessed the treatment outcome on day 7 as “major improvement” in 30% of the EPs 7630 group compared with 5.8% in the placebo group (P<.0001). Adverse events were reported in 6 patients in the EPs 7630 group (11.8%) and 2 in the placebo group (3.8%). In 4 cases in the EPs 7630 group, a causal relationship to the medication could not be excluded (GI complaints [3x], allergic skin reaction [1x]).
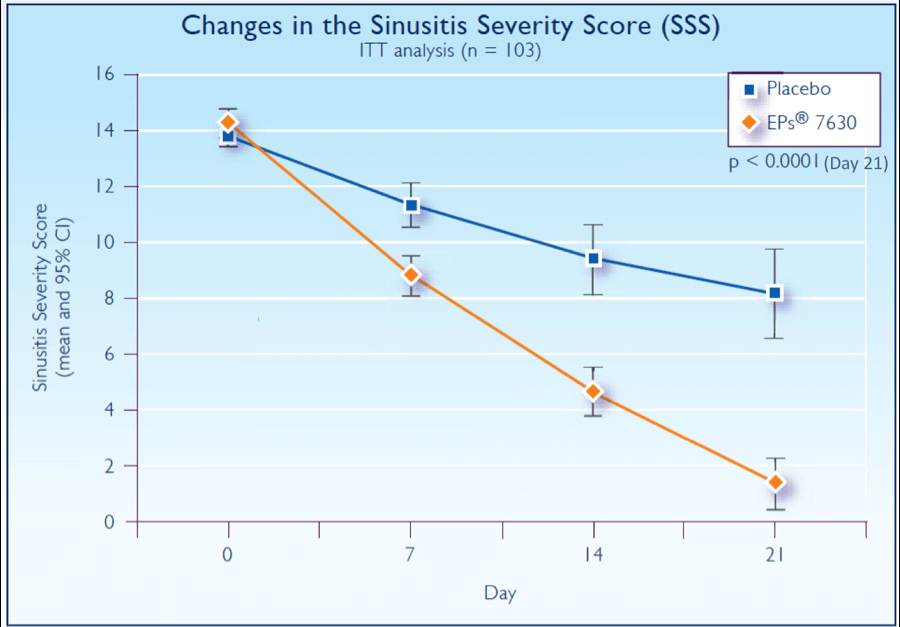
Figure 6.
Common Cold
Using a liquid extract of P sidoides (EPs) closely related to EPs 7630, a randomized, placebo-controlled study determined the efficacy of the extract for the treatment of the common cold.20 The study included 103 adult patients (18-55 y) with at least 2 major and 1 minor or with 1 major and 3 minor cold symptoms for 24 to 48 hours. Major cold symptoms included nasal discharge and sore throat; minor symptoms included nasal congestion, sneezing, scratchy throat, hoarseness, cough, headache, muscle aches, and fever.
The primary outcome measure was the Sum of Symptom Intensity Differences (SSID) of the Cold Intensity Score (CIS) from day 1 to day 5. The CIS consists of 10 symptoms considered to be associated with the common cold and designates them as major or minor (see above). At each patient visit, all symptoms except fever were rated according to a 5-point verbal rating scale with 0 meaning no symptoms to 4 being very severe. The maximum CIS score was 40 points. Secondary outcome criteria included diverse response according to the total CIS, changes of individual symptoms of the CIS, changes of further cold-related symptoms, ability to work, activity level, general well-being, health-related quality of life, time until onset of treatment effect, treatment outcome according to an integrative medicine outcome scale, and satisfaction with treatment according to the integrative medicine scale. After enrollment, patients were seen on days 3, 5, and 10.
From baseline to day 5, the mean SSID improved by 14.6±5.3 points in the EPs group compared with 7.6±7.5 points in the placebo group (P<.0001). The mean CIS decreased by 10.4±3.0 points and 5.6±4.3 points in the EPs and placebo groups, respectively (Figure 7). After 10 days, 78.8% of the EPs group was clinically cured (CIS=0 points or complete resolution of all but a maximum of 1 cold symptom) compared with 31.4% in the placebo group (P<.0001). The mean duration of inability to work was significantly lower in the EPs group (6.9±1.8 days) compared with the placebo group (8.2±2.1 days; P=.0003). Adverse events were recorded in 2/52 of the EPs group and 1/51 of the placebo group and classified as nonserious. A causal relationship to EPs could not be excluded in 1 EPs patient (epistaxis).
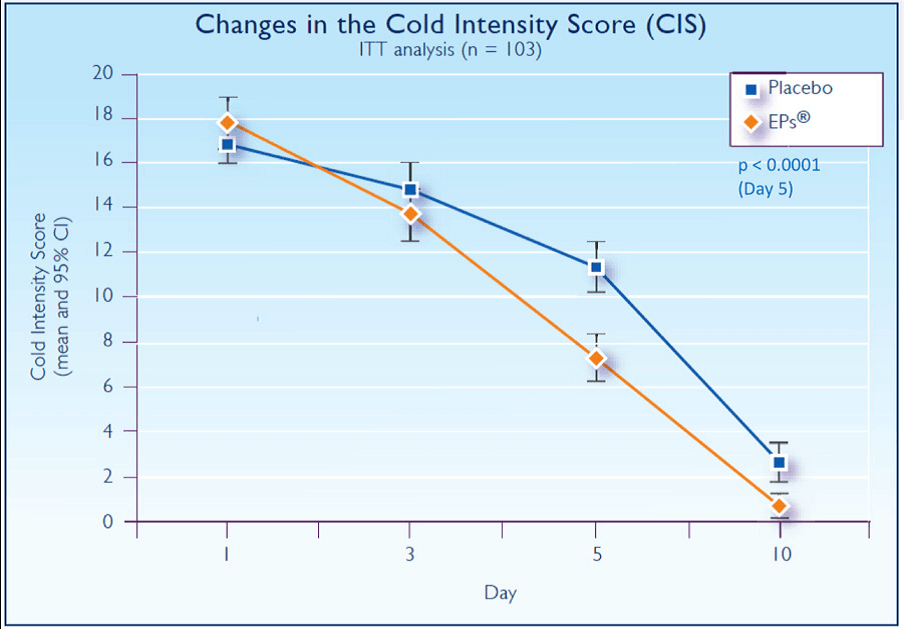
Figure 7.
Recommended Use and Safety
The primary indication for EPs 7630 is the treatment of acute, uncomplicated upper respiratory tract infections. The recommended dose of EPs 7630 for adults and children over the age of 12 years is 1.5 mL three times per day for 7 to 10 days. Children ages 6 to 12 years may take 1.0 mL three times per day. For children 2 to 5 years of age, the dose is 0.5 mL three times per day. While the data are preliminary, the currently recommended dose for the treatment of acute sinusitis in adults is 3.0 mL three times per day for 21 days.
Due to a lack of safety data, EPs 7630 is contraindicated during pregnancy and lactation. There are no known contraindications to concomitant use of EPs 7630 with other medications. Side effects are rare and have consisted primarily of mild gastrointestinal upset or skin rash.
Approximately 304 million daily doses of EPs 7630 were sold between 1994 and 2006, predominantly in Germany. The incidence of side effects is extremely low: 0.53 per million defined daily doses (DDD).21 This means that only 1 in 189,000 patients will experience a side effect during an average treatment period of 10 days. The rate of side effects is 0.27 per million DDD for hypersensitivity reactions (especially redness and pruritus), 0.13 for gastrointestinal disorders, and 0.05 for gingival hemorrhaging and nosebleeds.
One hundred sixty-one suspected cases of adverse drug reactions (ADRs) were reported spontaneously and 96 ADRs were reported from clinical trials between 1994 and 2006. The principal side effect from both sources was mild gastrointestinal upset.
Finally, in addition to randomized, placebo-controlled clinical trials, pediatric safety has been demonstrated in large postmarketing surveillance studies22,23 as well as the pharmacovigilance data listed above.
Conflict of Interest Disclosure
The author of this review paper is employed by Schwabe North America and its affiliates Integrative Therapeutics, Nature’s Way, and Enzymatic Therapy. The ingredient reviewed is contained in products sold by Integrative Therapeutics and Nature’s Way.
